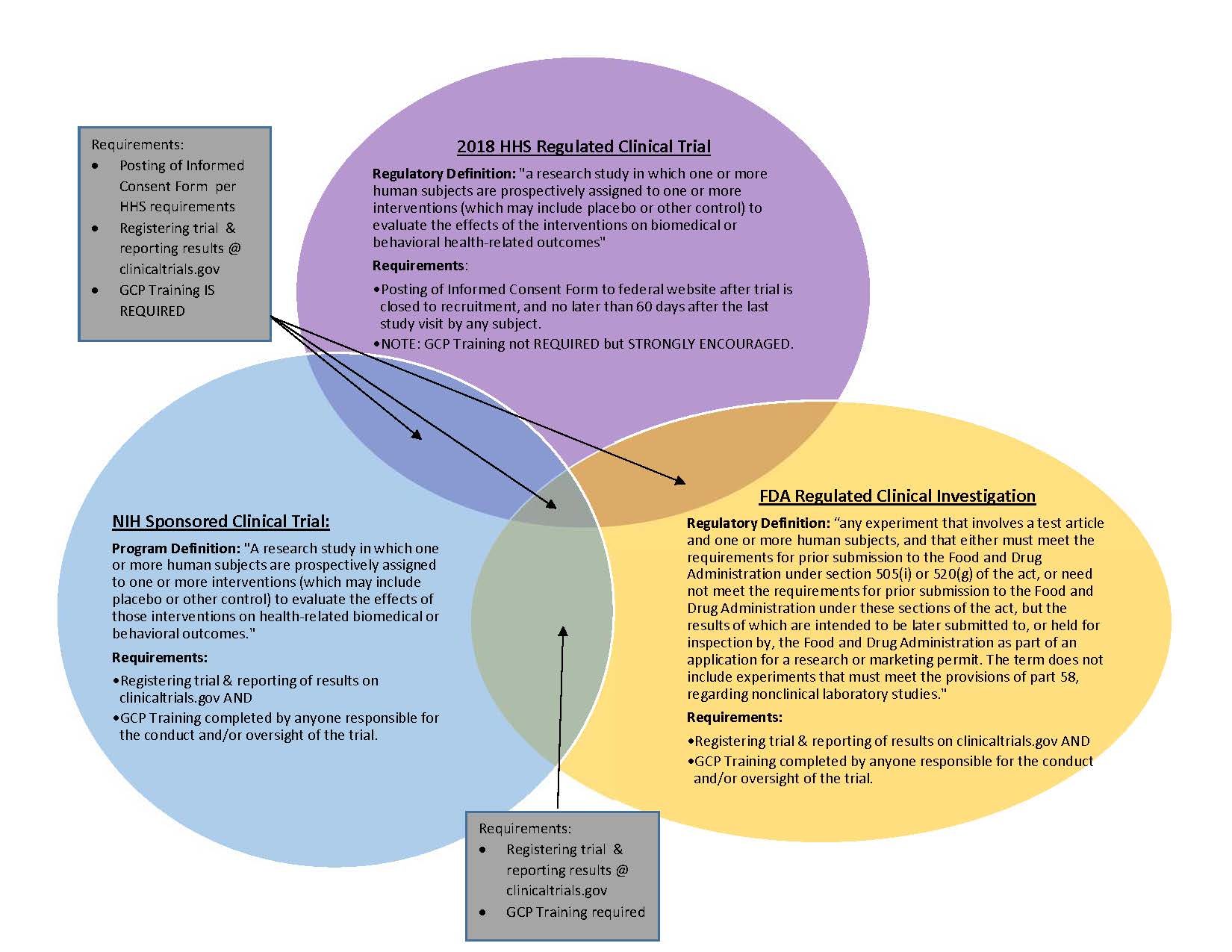
Some types of studies involving human subjects may be considered a clinical trial by regulatory definition and/or by funder/sponsor definitions. It is important to understand when a study is a clinical trial under these definitions as there are additional requirements that the Principal Investigator and research team must satisfy. Failure to satisfy these requirements in some circumstances may result in hefty fines for the investigator and/or institution and could impact future funding opportunities.
The most common definitions that apply to research at the University of Oregon are for studies regulated under the 2018 Revised Common Rule (U.S. Health and Human Services (HHS)), regulated by the Food and Drug Administration (FDA), and/or sponsored by National Institutes for Health (NIH). Each entity has differing requirements for clinical trials that meet their definition. At times, a study may meet multiple definitions and must comply with multiple sets of requirements.
Below are the definitions and requirements for these three entities; however, investigators need to be aware that other study funders/sponsors may have their own definitions and requirements. Investigators are responsible for reviewing the conditions of their award (e.g., Department of Defense) to determine if additional requirements are necessary.
Good Clinical Practice (GCP) and Training
Good Clinical Practice (GCP) is a set of broad regulatory requirements, standards, and recommendations that apply specific tasks, processes, and roles in the conduct of clinical research. Many of the standards used in the conduct of clinical trials are "best practices" derived from regulations, guidance, and industry standards and practices and not all found in black and white in the regulations. The Principal Investigator of the study is responsible for integrating GCP in the conduct of the research and the culture of the research team.
GCP training is a requirement for many regulated and/or federal sponsored clinical trials and investigations. However, GCP training offers researchers a foundation in best practices that can be applied in most research contexts. While GCP training is encouraged for any clinical trials/investigations conducted by UO researchers, many regulations and sponsors/funding agencies have specific requirements for completing, documenting, and maintaining GCP training. See sections below for the requirements associated with FDA regulated and NIH sponsored clinical research; however, investigators should be aware that other sponsors and entities may also require GCP training. Many entities have specific GCP training logs to support the logging, tracking and content of GCP training. Contact RCS for sample logs.
Training in GCP may be achieved through a class or course, academic training program, or certification from a recognized clinical research professional organization. NIH offers GCP training and UO offers online Good Clinical Practices (GCP) training through the Collaborative Institutional Training Initiative (CITI). CITI’s GCP courses are acceptable GCP training for the NIH policy. Learners will receive a completion report documenting the successful completion of CITI Program training.
HHS Clinical Trials
HHS regulations were revised in 2018 to include requirements for clinical trials governed by the revised regulations. Studies that remain governed under the pre-2018 regulations are not subject to these same requirements. Any studies reviewed under the 2018 regulations (either newly reviewed or transitioned oversight) are subject to the requirements below if they meet the HHS definition of clinical trial. Studies under the 2018 common rule that are not federally funded or supported do not generally need to comply with the HHS clinical trial requirements unless otherwise required by the IRB. However, they may also meet other definitions of clinical trials. Researchers can also voluntarily comply with the requirements.
HHS Definition of Clinical Trial: A research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of the interventions on biomedical or behavioral health-related outcomes.
HHS Requirements:
- For each clinical trial conducted or supported by a Federal department or agency, one IRB-approved informed consent form used to enroll subjects must be posted by the awardee or the Federal department or agency component conducting the trial on a publicly available Federal Web site that will be established as a repository for such informed consent forms.
- If the Federal department or agency supporting or conducting the clinical trial determines that certain information should not be made publicly available on a Federal Web site (e.g. confidential commercial information), such Federal department or agency may permit or require redactions to the information posted.
- The informed consent form must be posted on the Federal Web site after the clinical trial is closed to recruitment, and no later than 60 days after the last study visit by any subject, as required by the protocol.
How to Comply with the Requirements: At this time, two publicly available federal websites that will satisfy the consent form posting requirement, as required by the revised Common Rule, have been identified: ClinicalTrials.gov and a docket folder on Regulations.gov (Docket ID: HHS-OPHS-2018-0021). HHS and other Common Rule departments and agencies are developing instructions and other materials providing more information to the regulated community about this posting requirement.
It is the responsibility of the investigator to create a system for their research to ensure compliance with the posting requirement within the timeframe established by HHS regulations. Investigators are also responsible for determining any sponsor-specific instructions and other materials required related to the informed consent posting requirement.
FDA Clinical Investigations
Studies that meet the FDA definition of clinical trial and are also applicable drug or applicable device trials per FDA definitions, must comply with the FDA clinical trial requirements. A study can still be an applicable clinical drug or device study even if the study is IND/IDE exempt.
FDA Definitions
Clinical Trial means a clinical investigation or a clinical study in which human subject(s) are prospectively assigned, according to a protocol, to one or more interventions (or no intervention) to evaluate the effect(s) of the intervention(s) on biomedical or health-related outcomes.
Clinical investigation means any experiment that involves a test article and one or more human subjects and that either is subject to requirements for prior submission to the Food and Drug Administration under section 505(i) or 520(g) of the act, or is not subject to requirements for prior submission to the Food and Drug Administration under these sections of the act, but the results of which are intended to be submitted later to, or held for inspection by, the Food and Drug Administration as part of an application for a research or marketing permit. The term does not include experiments that are subject to the provisions of part 58 of this chapter, regarding nonclinical laboratory studies.
Applicable Clinical Trials: The FDA defines an Applicable Clinical Trial (ACT) as follows: (1) Trials of drugs and biologics: controlled clinical investigations, other than Phase 1 investigations, of a product subject to FDA regulation; and (2) Trials of biomedical devices: controlled trials with health outcomes of devices subject to FDA regulation, other than small feasibility studies, and pediatric post-market surveillance. The FDA has further elaborated the definition of an ACT which can be found here. For more resources, also see the ACT checklist and ACT FAQs. The Applicable Clinical Trial (ACT) definitions specific to drugs and devices is also included below.
- Applicable device clinical trial: (1) a prospective clinical study of health outcomes comparing an intervention with a device product subject to section 510(k), 515, or 520(m) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) (21 U.S.C. 360(k), 21 U.S.C. 360e, 21 U.S.C. 360j(m)) against a control in human subjects (other than a small clinical trial to determine the feasibility of a device product, or a clinical trial to test prototype device products where the primary outcome measure relates to feasibility and not to health outcomes); (2) a pediatric postmarket surveillance of a device product as required under section 522 of the FD&C Act (21 U.S.C. 3601); or (3) a clinical trial of a combination product with a device primary mode of action under 21 CFR Part 3, provided that it meets all other criteria of the definition under this part. [Source: 42 CFR 11.10(a); 81 FR 65139]
- Applicable drug clinical trial: a controlled clinical investigation, other than a phase 1 clinical investigation, of a drug product subject to section 505 of the FD&C Act (21 U.S.C. 355) or a biological product subject to section 351 of the Public Health Service Act (PHS Act) (42 U.S.C. 262), where “clinical investigation” has the meaning given in 21 CFR 312.3 and “phase 1” has the meaning given in 21 CFR 312.21. A clinical trial of a combination product with a drug primary mode of action under 21 CFR Part 3 is also an applicable drug clinical trial, provided that it meets all other criteria of the definition under this part. [Source: 42 CFR 11.10(a); 81 FR 65139]
FDA Requirements:
- GCP training: Must be completed, logged and tracked for clinical investigators and staff who are involved in the design, conduct, oversight, or management of the clinical trial. Recipients of GCP training are expected to retain documentation of their training. GCP training should be refreshed at least every three years in order to stay up to date with regulations, standards, and guidelines.
- Registering & reporting of results at clinicaltrials.gov: All ACTs are expected to register and submit results information to Clinicaltrials.gov, as per the Food and Drug Administration Amendments Act of 2017 (FDAAA). Applicable clinical trials are required to be registered in ClinicalTrials.gov not later than 21 calendar days after the enrollment of the first participant. Results information from those trials generally must be submitted not later than one year after the trial's primary completion date.
- Protocol Template: Applicants conducting phase 2 or 3 clinical trials that require Investigational New Drug applications (IND) or Investigational Device Exemption (IDE) applications can use a NIH-FDA template with instructional and sample text to help write protocols. Note that the use of the tool is voluntary and is not required for NIH applications or contract proposals. More information can be found on the NIH website.
- The following or similar information is provided to subjects or the subject LAR via the informed consent document: A description of this clinical trial will be available on http://www.ClinicalTrials.gov, as required by U.S. Law. This Web site will not include information that can identify you. At most, the Web site will include a summary of the results. You can search this Web site at any time.
-
- NOTE: If the study also falls under the HHS 2018 Revised Common Rule, a copy of the consent form must be posted to clinicaltrials.gov within the timeframe outlined in the HHS regulations as noted above.
How to Comply with the Requirements:
For detailed instructions and a comprehensive overview of how to comply with FDA requirements, use the links below.
- https://www.fda.gov/science-research/clinical-trials-and-human-subject-protection/fdas-role-clinicaltrialsgov-information
- https://clinicaltrials.gov/ct2/manage-recs/fdaaa
NIH Clinical Trials
NIH has a broad definition of clinical trial and often includes behavioral, interventional, mechanistic, exploratory and pilot research. Use the NIH Decision Tool and associated information on the NIH Clinical Trials website to evaluate if your study meets the definition of clinical trial as noted below. If a study is not funded or supported by the NIH, it is not in the scope of the NIH Policy on the Dissemination of Clinical Trial Information (NOT-OD-16-149). However, it may meet other definitions of a clinical trial.
NIH Definition of Clinical Trial: A research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes.
NIH Requirements:
- GCP training: must be logged and tracked for clinical investigators and staff who are involved in the design, conduct, oversight, or management of the clinical trial. Recipients of GCP training are expected to retain documentation of their training. GCP training should be refreshed at least every three years in order to stay up to date with regulations, standards, and guidelines. For more information, see the NIH FAQs for GCP requirements.
- Registering & reporting of results at clinicaltrials.gov: All NIH-funded clinical trials are expected to register and submit results information to Clinicaltrials.gov, as per the "NIH Policy on Dissemination of NIH-Funded Clinical Trial Information" for competing applications and contract proposals submitted on or after 1/18/2017. Applicable clinical trials are required to be registered in ClinicalTrials.gov not later than 21 calendar days after the enrollment of the first participant. Results information from those trials generally must be submitted not later than one year after the trial's primary completion date.
- Single IRB Requirements: For applicable domestic, multi-site clinical trials, the investigator must provide plans for use of a single Institutional Review Board (sIRB) to conduct the ethical review for human subjects in research.
- Protocol Template: Applicants conducting phase 2 or 3 clinical trials that require Investigational New Drug applications (IND) or Investigational Device Exemption (IDE) applications can use a NIH-FDA template with instructional and sample text to help write protocols. Note that the use of the tool is voluntary and is not required for NIH applications or contract proposals. More information can be found on the NIH website at https://grants.nih.gov/policy/clinical-trials/protocol-template.htm/.
- Consent Form Requirements
- The following or similar information is provided to subjects or the subject LAR via the informed consent document: A description of this clinical trial will be available on http://www.ClinicalTrials.gov, as required by U.S. Law. This Web site will not include information that can identify you. At most, the Web site will include a summary of the results. You can search this Web site at any time.
- NOTE: If the study also falls under the HHS 2018 Revised Common Rule, a copy of the consent form must be posted to clinicaltrials.gov within the timeframe outlined in the HHS regulations as noted above.
How to Comply with the Requirements: See NIH’s “Steps to Compliance for NIH Awardees” for more comprehensive information. This includes information on the following:
- Funding Opportunity Announcement (FOA) that accepts clinical trials and distinct review criteria for clinical trials.
- Required plan for the dissemination of NIH-funded clinical trial information that will address how the expectations of the NIH clinical trials policy.
NOTE: Failure to fulfill the NIH requirements for clinical trials may results in NIH withholding future funding to individual investigators and/or the institution.
ICMJE Publication Requirements for Clinical Trials
If you are interested in publishing results in a journal that follows the International Committee of Medical Journal Editors (ICMJE) policy and meet the ICMJE definition of a clinical trial, your study will require public registration of all clinical trials as a condition of publication.
- In June 2007 the ICMJE adopted the WHO's definition of clinical trial: "any research study that prospectively assigns human participants or groups of humans to one or more health-related interventions to evaluate the effects on health outcomes."
- Health-related interventions include any intervention used to modify a biomedical or health-related outcome (for example, drugs, surgical procedures, devices, behavioral treatments, dietary interventions, and process-of-care changes).
- Health outcomes include any biomedical or health-related measures obtained in patients or participants, including pharmacokinetic measures and adverse events.
- Purely observational studies (those in which the assignment of the medical intervention is not at the discretion of the investigator) will not require registration.
- The ICMJE member journals implemented the expanded definition of clinically directive trials for all trials that began enrollment on or after 1 July 2008. Those who are uncertain whether their trial meets the expanded ICMJE definition should err on the side of registration if they wish to seek publication in an ICMJE journal.
- ICMJE requires, and recommends that all medical journal editors require, registration of clinical trials in a public trials registry at or before the time of first participant enrollment as a condition of consideration for publication
For more information, see the ICMJE Clinical Trial Registration Policy, and FAQ "What is the ICMJE definition of a clinical trial?"
If you have questions, please contact Research Compliance Services.